Amplicon Pro generates full-length raw reads that correspond to intact amplicon molecules. By using Oxford Nanopore’s latest library preparation chemistry and R10.4.1 flow cell technology, this service enables accurate characterization of complex amplicon mixtures. Amplicon Pro is well suited for applications requiring resolution of heterogeneous amplicon populations, including pooled amplicons, variant libraries, and mixed PCR products. Multiple distinct consensus sequences can be generated per sample.
Our library preparation workflow begins with end-repair of amplicon DNA to ensure compatibility with ligation-based barcoding. Barcodes are then attached to the DNA ends using Oxford Nanopore's ligation-based chemistry, allowing samples to be multiplexed and sequenced on an Oxford Nanopore R10.4.1 flow cell.
After sequencing is complete, raw reads are basecalled using the latest Dorado Super Accuracy model. These high-accuracy reads are processed through our analysis pipeline, where they are grouped into multiple high-confidence consensus sequences, each representing a distinct amplicon population within the sample.
Once processing is complete, all results are uploaded to our secure customer portal, and you will receive an email notification when your data is ready. Details of all deliverables are outlined below.
Sequence Files
· FASTA file
A text-based file containing the final consensus DNA sequence.
· GenBank file
Includes the consensus sequence along with annotated genetic features.
· AB1 file
A Sanger-style chromatogram view of the consensus sequence, allowing visualization of variants and low-confidence positions. Each peak color corresponds to a specific nucleotide.
· Multi-FASTA file
A combined FASTA file containing all consensus sequences generated from the same sample.
Read-Level Data
· Per-base data file
Provides a detailed breakdown of how individual raw reads support each base in the consensus sequence. This file is used to generate the AB1 chromatogram.
· Alignment file
Displays the alignment of raw reads to the consensus sequence, enabling evaluation of sequencing data at a molecular level.
· FASTQ file
Contains all raw sequencing reads generated for your sample.
Report(s)
· Variant summary file
A table summarizing all detected variants, including the number of reads assigned to each variant and the corresponding sequence length.
· QC report file
A comprehensive quality control report containing key metrics and visualizations, including:
1. Read-length histogram
Displays the distribution of raw read lengths. A single dominant peak typically indicates a pure sample, while multiple peaks may suggest concatemers or additional DNA species.
2. Virtual gel
A simulated gel image generated from raw read data, providing an intuitive view of sample composition.
3. Additional sample metrics
Including total reads, total bases, reads mapped to consensus, sample name, consensus length, and related statistics.
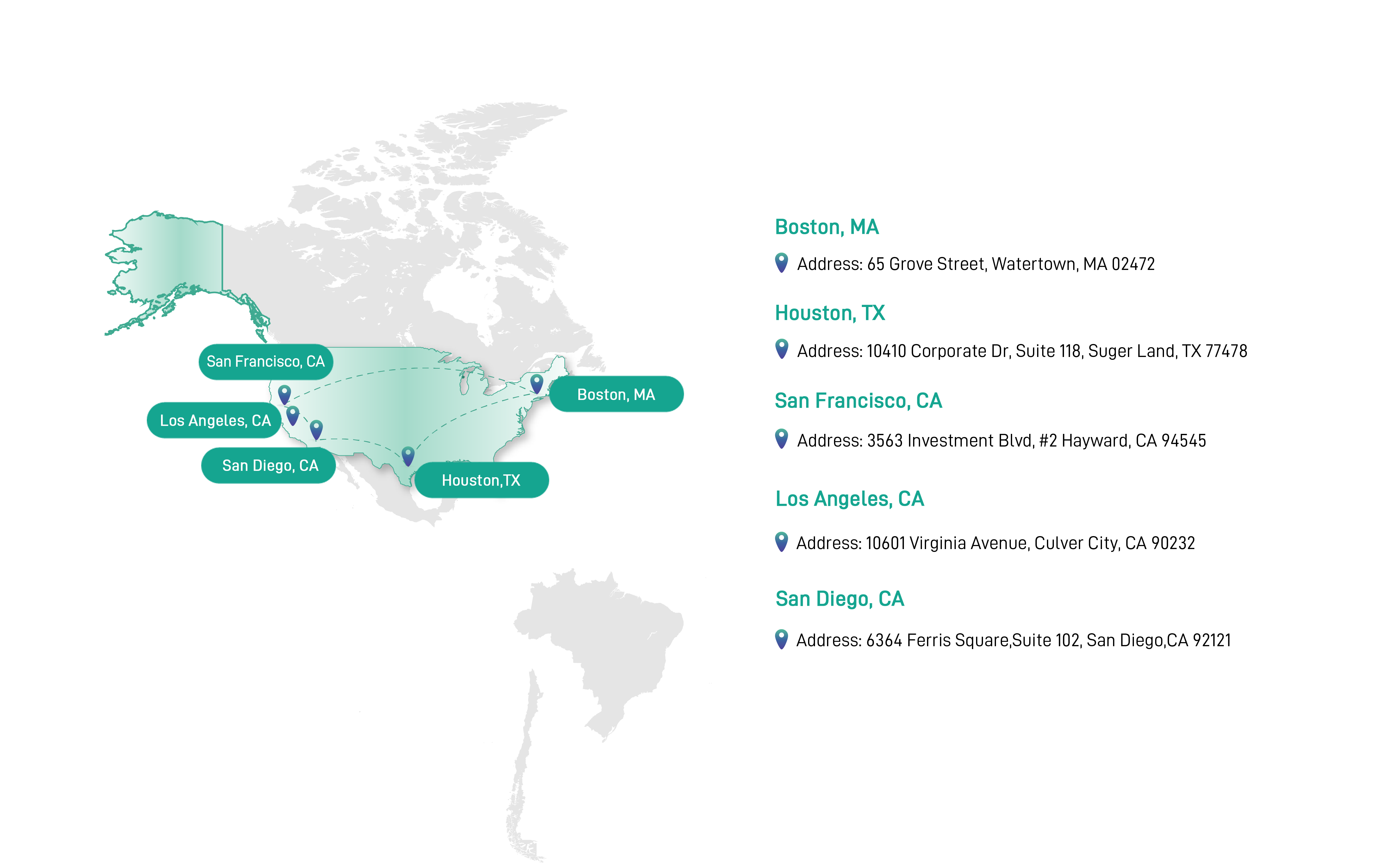
We Go Fast, You Go Forward
![]() sales.us@quintarabio.com
sales.us@quintarabio.com
Quintara Biosciences © 2025 All Rights Reserved | Site Map | Terms of Use | Privacy Policy